
Патологии, передаваемые по наследству обычно связаны с различными генными аномалиями. От них нельзя избавиться и остается лишь купировать возникающие симптомы.
Одной из таких болезней является дистрофический тип миотонии Россолимо Штейнерта Куршмана. Такое заболевание представляет собой дистрофию мышечных волокон на фоне развития миотонии.
Вместе с этим уменьшаются умственные способности, возникают эндокринные сбои и сбивается сердечный ритм.
Миотония — это болезнь, передаваемая по наследству, для которой свойственно замедленное расслабление мышц после их сокращения. Такое явление называется миотоническим спазмом. Узнают о наличии проблемы по свойственной симптоматике, а подтверждается она с помощью генеалогического анализа.
Особенности патологии
Впервые об этой болезни написал Г.И. Россолимо в начала ХХ века. Со временем в описание патологии вносились изменения, и она получила название дистрофическая миотония Россолимо Штейнерта Куршмана в честь людей, которые смогли ее изучить. Передается это заболевания аутосомно-доминантным методом, то есть от мамы или папы к малышу. Шансы на развитие болезни составляют 50%, если у одного из родителей есть аномальный ген, провоцирующий ее появление.
Страдает от этой патологии как женский, так и мужской пол. По статистике заболевают ею 10 человек на 200-300 тыс. населения. Впервые может проявиться дистрофический вид миотонии в раннем возрасте (до 10 лет), но зачастую ее признаки наблюдаются ближе к 20 годам.
Ученые выявили неприятную закономерность этого заболевания. Ее суть состоит в том, что если аномальный ген был у матери, то патология имеет значительно менее благоприятный прогноз. В случае передачи мутации от отца, течение болезни будет не таким тяжелым.
Причины
Все проблема дистрофической миотонии Россолимо Штейнерта Куршмана заключается в дефектном гене DMPK. Он локализован в 19 паре хромосом и в его функции входит выработка миотонин-протеинкиназы. В нормальном состоянии этот белок находится в скелетной мышечной ткани, нервной системе и составляющих миокарда.
Именно по этой причине его недостаточный синтез влияет на развитие различных осложнений свойственных этому патологическому процессу. Нехватка миотонин-протеинкиназы ведет к развитию миотонических спазмов и дистрофии мышечных тканей.
На фоне гипертрофии одних групп мышц, другие атрофируются и постепенно заменяются жировой и соединительной тканью.
У больных страдающих от миотонии дистрофического типа растет количество тринуклеотидных CTG-повторов. Они представляют собой основу мутировавшего гена. Количество тринуклеотидных CTG-повторов влияет на тяжесть болезни, а именно:
- Норма. У здоровых людей показатель CTG-повторов не превышает 37;
- Легкая форма. Если CTG-повторы находятся в рамках от 50 до 80, то диагностируется ранний этап болезни;
- Тяжелая форма. Для нее свойственен показатель повторов от 100 до 500;
- Врожденная форма. Диагностируют патологию у детей при 500-2000 CTG-повторов и возникает она преимущественно при передаче мутировавшего гена от матери.
Симптомы
В основном при дистрофической миотонии Россолимо Штейнерта Куршмана неврожденной формы первые проявления становятся заметны примерно от 6 до 35 лет. Однако зачастую симптоматика становится ярко выраженный к 20 годам. Проявления миотонии сочетаются с миопатией. Она представляет собой первичное дистрофическое поражение нервных тканей. Зачастую болезнь протекает вместе с поражением нервной и сердечно-сосудистой системы, а также ей свойственны эндокринные сбои и развитие катаракты.
Наиболее характерным признаком дистрофической миотонии являются миотонические спазмы. Больше всего им подвергаются сгибатели кисти и мышечные ткани, отвечающие за жевание.
У больных также появляется реакция свойственная миотонии на механическое воздействие, например, специального молоточка. Обычно это заметно во время осмотра у невролога.
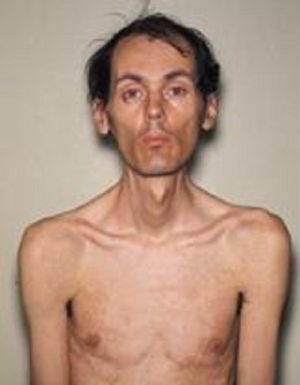
Особым признаком миотонии Россолимо-Штейнерта-Куршмана является постепенное угасание миотонических проявлений на фоне усугубление дистрофических изменений. Зачастую подвергаются атрофии мышцы отдаленных отделов нижних и верхних конечностей, а также лицевые, височные и грудино-ключично-сосцевидные мышечные ткани.
Пораженная мимика лица обычно проявляется в виде печального выражения лица. При дистрофических изменениях мышечной ткани глотки и гортани у больного меняется голос и ему становится трудно глотать.
Одним из наиболее неприятных проявлений дистрофической миотонии Россолимо Штейнерта Куршмана является влияние патологии на дыхательные мышечные ткани. Больные при этом мучаются от припадков апноэ (остановки дыхания) во время сна и пневмонии застойного и аспирационного типа.
Дисфункции сердечно-сосудистой системы наблюдаются у каждого второго человека, страдающего от дистрофической миотонии. Больные чаще всего страдают от сбоев в сердечном ритме, возникшем на фоне нарушения проводимости и увеличения левого желудочка. Не реже встречается и другой патологический процесс, а именно блокада ножек пучка Гиса.
Среди симптомов повреждения центральной нервной системы можно выделить стремительное уменьшение умственных способностей (вплоть до легкой дебильности) и сбои в ритме сна. Люди, страдающие от миотонии, начинают дольше спать и все равно не высыпаются.
Свойственные дистрофической миотонии Россолимо Штейнерта Куршмана эндокринные нарушения преимущественно касаются лишь половой сферы. У мужского пола это выражается следующей симптоматикой:
- Уменьшение либидо (тяги к противоположному полу);
- Задержка в опускании или отсутствие одного из яичек;
- Недостаточная выработка половых гормонов;
- Импотенция;
- Дисфункция половых желез.
У женского пола признаки эндокринных сбоев выглядят следующим образом:
- Рост волос по мужскому типу;
- Сбои в менструальном цикле;
- Развитие раннего климакса.
У обоих полов наблюдается алопеция (облысение). У мужчин она возникает преимущественно в области лба и висков, а у женщины проблема имеет диффузный характер или имеет очаговое проявление.
Признаки врожденной дистрофической миопатии
Заметить врожденный вид патологии можно еще во время внутриутробного развития. Обычно врач выявляет недостаточную активность плода. Для диагностики гинеколог использует ультразвуковое обследование (УЗИ). Выявить эту проблему можно после 6 месяца беременности.
Родившийся ребенок страдает преимущественно от проявлений миопатии, а именно от пониженного мышечного тонуса. Проблема касается в основном мышц конечностей, а также мимической, глазодвигательной и жевательной мускулатуры. Из-за этого часто наблюдаются проблемы при кормлении. Проявления миотонии становятся заметны значительно позже.
Дети, рожденные с дистрофической миотонией, мучаются от замедленного моторного развития и олигофрении. Симптоматика у них прогрессирует достаточно быстро, поэтому летальный исход часто возникает еще совсем в юном возрасте.
Диагностика
Заподозрить наличие проблемы невролог может по нестандартному сочетанию признаков свойственных болезни Россолимо Штейнерта Куршмана. Для подтверждения или опровержения диагноза врач назначит генеалогический тест и исследует ДНК код больного. Дополнительно может потребоваться проведение инструментальных методов обследования:
- ЭКГ;
- Электромиография;
- Анализ половых гормонов;
- Электронейрография.
Для точного дифференцирования диагноза невролог часто направляет больного к таким врачам
- Кардиолог;
- Андролог (мужчины);
- Гинеколог (женщины);
- Генетик;
- Эндокринолог.
Во время диагностики врач должен будет дифференцировать патологию Россолимо Штейнерта Куршмана среди прочих диагнозов, например, от патологии Томсена, которая также проявляется мышечной гипертрофией. Это же относится и к болезни Беккера, так как заболевания отличаются лишь скоростью поражения лицевой мышечной ткани и типом наследования.
Курс терапии
Такая болезнь, как миотония дистрофического типа Россолимо Штейнерта Куршмана на сегодняшний день неизлечима. Людям, страдающим от нее, приходится сидеть на строгой диете, в которой не должно быть калия. Помимо изменения рациона, врачи советуют больным беречь себя от переохлаждения, так как холод провоцирует миотонический спазм.
Медикаментозная терапия служит для замедления течения болезни и предотвращения приступов миотонических спазмов. Обычно используют такие препараты:
- Новокаимид;
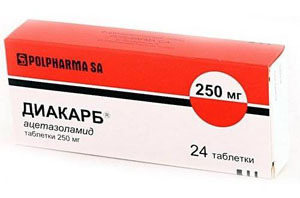
- Диакарб;
- Хинин;
- Дифенин.
Вместе с ними, больные должны употреблять анаболики по типу Неробола и Метиландростендиола для замедления дистрофических изменений. В обязательную схему лечения также должен входить АТФ (аденозин-трифосфатогистидинато-магний) и витаминные комплексы.
Благодаря такому курсу терапии снизится интенсивность проявлений болезни и улучшится состояние больного. Вылечить миотонию такие методы не смогут, но они продлят жизнь, а это не менее важный результат, так как со временем ученые придумывают все более эффективные лекарства.
Дистрофический вид миотонии является тяжелым и неизлечимым заболеванием, которое имеет неблагоприятный прогноз. При возникновении ее первых признаков необходимо срочно обратиться к врачу для прохождения диагностики и составления курса терапии.
Источник: http://NashiNervy.ru/perifericheskaya-nervnaya-sistema/simtomy-distroficheskoj-miotonii-rossolimo-shtejnerta-kurshmana.html
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Типичным представителем является миотоническая дистрофия (или дистрофическая миотония), описанная в начале прошлого века несколькими авторами и получившая название болезни Россолимо-Штейнерта-Куршмана.
Этот недуг является самым известным заболеванием из разряда миотоний и самой распространенной формой мышечной дистрофии у взрослых людей. Что представляет собой эта болезнь и как с ней бороться?
Открытие и суть заболевания
Россолимо, Штейнерт и Куршман изучали болезнь, являющуюся генетической патологией с аутосомно-доминантным типом наследования. Это значит, что один родитель имеет мутантный ген, больные дети при этом рождаются с вероятностью 50%. Заболевание носит характер семейного недуга и передается последующим поколениям по вертикали.
Сыновья и дочери в таких семьях болеют с одинаковой частотой, примерно 3 — 5 человек на 100 тысяч населения. Возраст начала заболевания, а также выраженность симптомов отличаются значительной вариабельностью.
В основе болезни лежит дефект гена из 19 пары хромосом, который отвечает за синтез фермента миотонин-протеинкиназы. Это белок в норме присутствует не только в скелетной мускулатуре, но и в клетках миокарда и ЦНС.
Вот почему для дистрофической миотонии характерна полисистемность проявлений с поражением разных органов и систем. Неполноценность миотонин-протеинкиназы приводит к появлению мышечных спазмов вместе с атрофическими изменениями мускулатуры головы, шейного отдела, конечностей.
Наблюдается сочетание гипертрофии одних мышечных волокон с атрофией других и заменой их на жировую или соединительную ткани.
Клинические проявления
В связи с варьированием начала заболевания в клинике различают следующие формы по возрастному принципу:
- врожденная форма — манифестация болезни начинается сразу после появления ребенка на свет;
- юношеский вариант — дебют миотонии в возрасте от одного года до периода полового созревания;
- классическая форма — начало клинических проявлений приходится на второй и третий десяток жизни;
- минимальный вариант — манифестация приходится на поздние сроки — шестой десяток жизни.
Характерно, что чем позднее проявляется болезнь, тем благоприятнее течение и лучше прогноз. Чаще всего встречается классическая форма болезни Штейнерта, для которой типичными являются следующие клинические симптомы:
- Миотония — проявляется спазмами жевательных мышц и сгибателей кистей рук, характерны атрофические изменения в разных группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии.
- Сердечнососудистые нарушения — нарушения ритма сердца, гипертрофические изменения левого желудочка, выявляемые на ЭКГ, застойная сердечная недостаточность.
- Эндокринные расстройства (в основном затрагиваются половые функции) — уменьшение размеров половых органов, снижение сексуального влечения, у женщин — расстройства менструального цикла, ожирение.
- Общие изменения дистрофического характера — сухость и пигментация кожных покровов, выпадение частично или полностью волос и зубов, ранняя катаракта.
- Нарушения со стороны ЦНС — усталость, расстройства сна, апатия, потеря интеллекта.
 группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии.
группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии.Отдельно стоит отметить характерные клинические проявления врожденной формы дистрофической миопатии:
- уменьшение активных движений плода в утробе матери, выявляемое во время УЗИ;
- в период новорожденности — вялость, распространенная гипотония, особенно в жевательных, мимических, мышцах глазных яблок;
- сохранение и даже повышение сухожильных рефлексов;
- проблемы вскармливания, расстройства дыхания по типу респираторного дистресс-синдрома;
- задержка физического и нервно-психического развития, признаки олигофрении;
- стремительное прогрессирование заболевания, высокий риск внезапной смерти.
Диагностические критерии
Подозрение на болезнь Россолимо-Штейнерта — Куршмана может возникнуть у врача при наличии у пациента сочетания миотонических и дистрофических изменений в мышцах на фоне потери интеллекта и наличия сердечнососудистой и эндокринной патологии.
Полисистемность практически всегда свидетельствует о генетической природе заболевания. Такие больные подлежат анализу ДНК и проведению генеалогического анализа для подтверждения аутосомно-доминантного наследования патологии. В качестве информативных методов исследования используются электрокардиография, электронейромиография, анализы на гормоны.
В связи с многогранностью клинических проявлений к процессу постановки диагноза обычно привлекаются специалисты из разных отраслей медицины — генетики, кардиологии, эндокринологии, гинекологии, андрологии, неврологии.
Дифференциальный диагноз проводится между дистрофической миотонией и другими видами схожих заболеваний. В отличие от остальных для болезни Россолимо характерна мышечная атрофия. Нередко для подтверждения диагноза приходится прибегать к биопсии, чтобы определить уровень мышечного белка, который в тканях при данной патологии повышен.
Проводится также антенатальная диагностика методом исследования околоплодных вод.
Медицинская помощь
Генетическое заболевание невозможно излечить полностью, поэтому целью лечения при болезни Россолимо-Штейнерта-Куршмана является купирование симптомов, улучшение общего состояния и социальная адаптация пациентов.
Принципы лечения заключаются в следующем:
- диета с низким содержанием солей калия (яблоки, спаржа, капуста, огурцы, виноград, зелень, кукуруза, ягоды, редис, мандарины, грейпфрут, лук, морковь, баклажаны, горох);
- исключение переохлаждений во избежание возникновения спазмов;
- применение препаратов хинина для стабилизации клеточных мембран, таких лекарств, как Дифенин, Новокаинамид, Диакарб — для снятия мышечных спазмов и уменьшения скованности, судорог, снижения внутричерепного давления;
- использование анаболических стероидов (Метанандростенолон, Ретаболил, Нерабол), витаминов группы В, АТФ для стимулирования мышечной массы;
- ЛФК, массаж, электромиостимуляция, ортопедические приспособления.
 грейпфрут, лук, морковь, баклажаны, горох);
грейпфрут, лук, морковь, баклажаны, горох);Перечисленные мероприятия дают хороший положительный эффект как при классической, так и при врожденной форме болезни. Полностью избавить больного от дистрофической миотонии они не могут, но продлить ему жизнь и улучшить ее качество способны.
В случае классической формы заболевание может протекать долго при проведении своевременных лечебно-коррекционных мероприятий. Наиболее благоприятный прогноз у поздно проявившихся форм болезни.
Профилактические мероприятия сводятся к тому, что женщине из семьи с неблагополучным анамнезом на стадии планирования беременности необходимо пройти обследование на наличие аномальных генов, ответственных за развитие мышечной дистрофии. Это целесообразно сделать также в случае наличия у родственников отца ребенка данной патологии.
Возможности к рождению деток должны решаться индивидуально в каждом конкретном случае врачами — генетиками после консилиума.
Читайте ещё
Источник: http://NeuroDoc.ru/bolezni/myshechnye/distroficheskaya-miotoniya-rossolimo-shtejnerta-kurshmana.html
Дистрофическая миотония: клиническая картина и лечение
Исследователи выделяют две группы миотонических синдромов:
- дистрофические;
- недистрофические.
В каждой группе существует своя дополнительная классификация патологий в зависимости от особенностей проявлений, времени развития.
Дистрофические миотонии
Болезни данной группы объединяются наследованием по доминантному принципу и характеризуются миотоническим, вегетативно-трофическим и дистрофическим синдромом. Особенностью их является отложенное расслабление после напряжения, растущая слабость мышц и их атрофия.
По времени возникновения говорят о конгенитальной, ювенильной, взрослой и поздней форме. Конгенитальная проявляется у ребенка сразу после рождения. Ювенильная – от года до подросткового периода. Взрослая – от 20 лет до 40. Поздняя развивается по достижении сорокалетнего возраста.
В зависимости от того, какой ген мутировал, выделяют дистрофическую миотонию I, II и III типа. К первому относят патологии, которые представляют собой форму, переходную между миотонией и миопатией. Типичный пример – болезнь Россолимо-Штейнерта-Куршмана. Поражаются мышцы конечностей, органов дыхания, миокарда. Уже в детском возрасте появляются нарушения скелета.
Патологии второго типа проявляются у людей разного возраста – от 7 лет до 60. Наблюдается скованность движений, сопровождаемая болями. По мере развития отмечается слабость мышц конечностей, кистей. Развиваются эндокринные заболевания.
Третьему типу свойственна слабость глубоких мышц конечностей, тела. Атрофия мускульной ткани шеи и плеч ведет к свисанию головы. Нарушается внимание, мышление, память.
Недистрофические миотонии
К этому типу относят патологии, связанные с изменением генов натриевого и хлорного каналов. Основным проявлением является слабость в кистях.
Миотонии натриевого канала включают калийзависимую, врожденную парамиотонию, а также гиперкалиемический периодический паралич с миотонией.
К калийзависимому типу относят патологии, передаваемые по рецессивному и доминантному признаку. Возникает этот тип миотонии у детей от 5 лет, взрослых до 55. Характеризуются спазмами, мышечными болями. Сильнее всего поражаются нижние конечности.
Врожденная парамиотония передается по доминантному признаку. Провоцирующим фактором является холод. Важной особенностью является преходящая слабость мышц, которая может длиться несколько дней. Страдают преимущественно жевательные и мимические мышцы.
Гиперкалиемический периодический паралич с миотонией проявляется до 10 лет, наследуется по доминантному признаку. Приступы слабости возникают после приема продуктов с большим содержанием калия. Возникают в ногах, распространяют на тело и руки. Длятся такие эпизоды до двух часов.
К каналопатиям хлорного канала относят миотонию Томсена и болезнь Беккера. Первой свойственно спазмирование мышц пальцев и жевательной мускулатуры. Проявляется в раннем возрасте, в некоторых случаях состояние стабилизируется. В целом мышечная ткань остается достаточно развитой.
Миотонию Беккера обнаруживают у детей от 4 лет до 18. Имеет более тяжелое течение по сравнению с болезнью Томсена. Характеризуется мышечными болями. Поражаются дистальные, мимические мышцы, мускулатура конечностей.
Миопатия Дюшенна: как продлить жизнь человека с генетическим заболеванием
Этиология
Все виды миотоний обусловлены генетическими нарушениями. В ряде случаев провоцирующим фактором является аутосомно-доминантная передача, в других – аутосомно-рецессивная.
Так, миотония Томпсона наследуется по доминантному принципу, т. е. ген передал ребенку один из его родителей. К этой группе относят также врожденную парамиотонию Эйленбурга, миотонию Россолимо-Штейнерта-Куршмана.
По рецессивному типу развивается болезнь Беккера, возникает она из-за передачи мутировавшего гена обоими родителями. Считается, что патологии этого типа проявляются в раннем детстве и имеют более тяжелое течение.
Мутированные гены вызывают нарушение проницаемости мембран клеток, изменения ионных каналов хлора и натрия, медиаторного обмена веществ, что, в конечном счете, ведет к нарушению функций мышечной ткани.
Механизм развития разных видов миотоний един. Ослабленная мускульная ткань из-за воздействия на нее тех или иных факторов приходит в сильный тонус. Возникает состояние, которое называют миотонической атакой. Она появляется в тот момент, когда человек пытается сделать движение, требующее вовлечения пораженных мышечных волокон.
Провоцирующими факторами могут быть стресс, холод, сильные эмоции, длительная обездвиженность.
Считается, что в некоторых ситуациях патологию вызывают кровнородственные связи.
Симптомы
Характерным признаком всех миотоний является симптом «кулака». Характеризуется он тем, что, сжав кулак, больной не может его быстро разжать. Для этого ему потребуется приложить определенные усилия. При последующих сжатиях кулак разжимается легче. Скованность возрастает только при миотонии Эйленберга.
Общие затруднения при всех формах заболевания возникают при попытке открыть рот, встать со стула, быстро открыть глаза, которые до того были закрыты.
Тяжесть проявления симптомов позволяет выделить легкую, среднетяжелую и тяжелую форму заболеваний. Последняя характерна преимущественно для врожденных заболеваний.
Миотония Томпсона и Беккера
В начале развития заболевания возникают болезненные спазмы мышц голени. В дальнейшем поражаются мышцы лица, глотки, языка. Симптомы могут уменьшаться с возрастом. У некоторых они и вовсе исчезают. На смену им приходят парезы и атрофия мышечных волокон головы, шеи. При уменьшении жевательных мышц происходит западение щек. Атрофия шейных волокон ведет к запрокидыванию головы.
Позже затрагиваются мышцы конечностей. Возрастает их слабость, уменьшается сила.
Страдает сердечно-сосудистая система. Возникают эпизоды аритмии, брадикардии, снижения артериального давления. Выпадают волосы, зубы, кожные покровы становятся очень тонкими.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Первые симптомы проявляются в возрасте 15-20 лет, иногда – в 35.
Появляются спазмы мышц, двигательная возбудимость, с развитием болезни эти признаки угасают, чего нельзя сказать о комплексе миопатических симптомов.
Развивается атрофия мышечной ткани лица, шеи, кистей рук, снижаются сухожильные рефлексы. Реже поражаются мышцы ног и висков. Постепенно нарастает слабость, больные жалуются на быструю утомляемость.
Атрофия мышц гортани ведет к нарушению глотания, осиплости или потере голоса. У мужчин развивается импотенция, у женщин нарушается менструальный цикл. Часто возникают нарушения сердечно-сосудистой деятельности, ведущие к аритмии, брадикардии.
У многих больных появляется катаракта. Во сне возможны приступы апноэ.
Болезнь Лейдена-Томсена-Беккера
Основной признак – невозможность расслабить мышцы после их напряжения, возникают спазмы. Они поражают человека, когда он закрывает глаза, смыкает челюсти или сжимает руки в кулак. При этом обратное движение в течение долгого времени сделать не получается.
По внешнему виду больные похожи на спортсменов. На ощупь мышцы твердые, плотные, однако силы в них нет.
Миотония хондродистрофической формы
Больных отличает низкий рост, врожденный вывих бедра, скованная мимика, скованность суставов.
Врожденная дистрофическая миотония
Патология характеризуется нарушением у ребенка частоты сердечного ритма, повышенной сонливостью, усилением скованности в холод, эндокринными патологиями.
Парамиотония Эйденбурга
Расслабление мышц затрудняется при холодной температуре окружающего воздуха или местного воздействия. Так, при употреблении сильно холодных продуктов спазм охватывает глотку и язык. Снимается он после согревания.
При общем переохлаждении возникает так называемый «холодный паралич».
Диагностика
Для определения точного диагноза проводится осмотр больного, проверка сухожильных рефлексов, собирается информация о ходе развития патологии, назначаются следующие исследования:
- Электромиография. Регистрируются биоэлектрические импульсы различных участков мышечной ткани, характеризующие поражения нервной системы. Проводится преимущественно электромиографическое исследование.
- ДНК-диагностика.
- Биохимический анализ крови. Обнаруживаются антитела к калиевым каналам, повышенное содержание креатинфосфокиназы.
- Гормональное исследование. Проводится при обнаружении эндокринных нарушениях.
- ЭКГ. Назначается для контроля возникновения и развития сердечно-сосудистых патологий.
Основной целью является дифференциальная диагностика миотонии одного вида от другого.
Лечение
В настоящее время проводится только симптоматическое лечение миотонии. Каких-либо способов полностью остановить течение болезни не существует.
Для снижения судорог и расслабления мышечных тканей назначается Фениотин, Дифенин, Мексилетин. С целью уменьшения содержания калия – диуретики. Подавляют иммунные реакции введением иммуноглобулина. При необходимости применяются анаболитические средства. Тяжелые случаи патологии лечат курсами глюкокортикоидов. Аритмию снимают Новокаинамидом, Хинином.
Некоторым больным назначают курсами препараты, направленные на улучшение обмена веществ (Актовегин), ноотропные лекарства, способные убрать последствия чрезмерного двигательного возбуждения (Пантогам).
Важную роль в предотвращении развития заболевания и облегчении его симптомов играет диета. Она основана на ограничении потребления продуктов, содержащих калий.
Назначается физиотерапия. Основной метод – электромиостимуляция, направленная на стимуляцию нервно-мышечной системы с помощью электрических импульсов.
Рекомендован массаж. Его проводят курсами по 2-3 раза в год.
Несколько раз в год проводится ЛФК с физиотерапевтом. В остальное время показано выполнять физические упражнения в домашних условиях. Рекомендуется плавать в бассейне. Физическая активность помогает нормализовать мышечный тонус, восстановить активность мышц.
Осложнения
Опасность для людей, страдающих миотонией, представляют последствия этой патологии. Среди них выделяется апноэ, пневмония, болезни сердца, аритмия, снижение интеллекта.
Прогноз
Легкие формы патологии не ведут к потере трудоспособности и летальному исходу. В случае развития осложнений, связанных с заболеваниями сердца, возможна смерть от его остановки.
Профилактика
Генетическая обусловленность миотоний не оставляет возможностей для ее профилактики. Единственная возможная мера – проведение ДНК-экспертизы перед планированием беременности. Рекомендована она, прежде всего, тем, чьи родственники страдают от этой патологии.
Миотония представляет собой группу разнородных заболеваний, характеризующихся спазмированием после их напряжения. Патологии носят прогрессирующий характер, однако редко приводят к инвалидизации и гибели. В настоящее время лечение направлено только на снятие остроты симптомов.
Для подготовки статьи использовались следующие источники:Латышева В. Я., Дривотинов Б. В., Олизарович М. В. // Неврология и нейрохирургия: учеб. пособие — Минск, Выш. шк. 2013.
Коллектив авторов // Нервные болезни — «СпецЛит», 2011 (Учебник для средних медицинских учебных заведений).Гусев Е. И., Коновалов А. Н., Скворцова В. И. // Неврология и нейрохирургия под ред. Коновалова А. Н.
, Козлова А. В. — 2014.
Источник: https://neuromed.online/miotoniya/
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
G71.1 Миотонические расстройства
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу.
Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет.
В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Последние исследования генетического набора больных дистрофической миотонией показали, что основу заболевания составляет дефект в гене DMPK, находящемся в 19-й хромосоме и отвечающем за синтез миотонин-протеинкиназы. У больных дистрофической миотонией выявляется значительное увеличение тринуклеотидных CTG-повторов в основной части гена DMPK. При этом именно от количества повторов зависит форма и тяжесть миотонии.
В норме число тринуклеотидных повторов варьирует от 5 до 37. Увеличение повторов до 50-80 приводит к появлению мягкой формы миотонии Россолимо-Штейнерта-Куршмана. Если количество тринуклеотидных повторов находится в промежутке от 100 до 500, развивается поздняя форма заболевания.
Врожденные формы дистрофической миотонии возникают при повышении числа CTG-повторов от 500 до 2000. Исследования показали, что увеличение тринуклеотидных повторов происходит в основном в женских гаметах в процессе мейоза.
В связи с этим при передаче заболевания от матери у ребенка возникает более тяжелая форма миотонии или ее врожденный вариант.
В классическом варианте миотония Россолимо-Штейнерта-Куршмана начинает проявляться после первых 5 лет жизни и может манифестировать до 35-летнего возраста.
Но наиболее часто клинические проявления заболевания возникают в возрастном диапазоне от 10 до 20 лет.
Они представляют собой сочетание типичных симптомов миотонии с признаками миопатии, поражением сердечно-сосудистой системы и ЦНС, эндокринными нарушениями и катарактой.
Из миотонических проявлений для миотонии Россолимо-Штейнерта-Куршмана характерны миотонические спазмы, наиболее выраженные в жевательных мышцах и мышцах-сгибателях кисти.
Наблюдаются также механические реакции миотонического типа, выявляемые при ударе неврологическим молоточком. Отличительной особенностью миотонии Россолимо-Штейнерта-Куршмана является наличие атрофических изменений в различных группах мышц.
При этом течение заболевания характеризуется постепенным угасанием симптомов миотонии на фоне прогрессирующей мышечной дистрофии.
Чаще всего при миотонии Россолимо-Штейнерта-Куршмана поражаются мышцы дистальных отделов конечностей, мимическая мускулатура, грудино-ключично-сосцевидные и височные мышцы. Поражение мимических мышц проявляется характерным маскообразным печальным выражением лица больных дистрофической миотонией.
Атрофические изменения мышц глотки и гортани приводят к развитию миопатического пареза гортани с нарушением голоса и затруднением глотания. Миопатические изменения могут возникать в дыхательной мускулатуре.
Наряду с миотоническими спазмами они приводят к ухудшению легочной вентиляции, появлению приступов апноэ во сне, возникновению застойной или аспирационной пневмонии.
Нарушения сердечно-сосудистой системы наблюдаются примерно в половине случаев дистрофической миотонии. К ним относятся аритмии, связанные с нарушением проводимости, и гипертрофия левого желудочка. Наиболее распространена блокада ножек пучка Гиса. Из признаков поражения ЦНС чаще всего наблюдается гиперсомния и снижение интеллектуальных способностей, доходящее до легкой степени дебильности.
Эндокринные расстройства при миотонии Россолимо-Штейнерта-Куршмана затрагивают в основном половую сферу.
У мужчин они проявляются снижением либидо, крипторхизмом, импотенцией, гипогонадизмом, у женщин — гирсутизмом, нарушениями менструального цикла (олигоменореей, дисменореей) и ранним климаксом.
Типичным является изменение структуры волос в сочетании с алопецией. У мужчин отмечается выпадение волос на висках и в области лба, у женщин — диффузное или очаговое облысение.
Первые признаки врожденной формы миотонии Россолимо-Штейнерта-Куршмана могут проявляться еще в период внутриутробного развития. Как правило, они выражаются в значительном снижении двигательной активности плода, которое диагностируется акушером-гинекологом по данным акушерского УЗИ в III триместре беременности.
После рождения ребенка преобладают симптомы миопатии. Отмечается диффузная гипотония мышц, более выраженная в мимической, жевательной и глазодвигательной мускулатуре, а также в мышечных группах дистальных отделов конечностей.
Характерны затруднения вскармливания и дыхательные расстройства. Миотоническая симптоматика начинает проявляться несколько позже. Врожденная дистрофическая миотония сопровождается задержкой моторного развития и олигофренией.
Типично быстрое прогрессирование симптомов заболевания, часто приводящее к смертельному исходу еще в раннем детстве.
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологу предположить миотонию Россолимо-Штейнерта-Куршмана.
Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ.
К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии.
Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия.
От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.
Радикальной терапии миотонии Россолимо-Штейнерта-Куршмана пока не существует. Пациентам, имеющим это заболевание, показана диета со сниженным содержанием калия. Им также следует избегать переохлаждения, которое провоцирует миотонические спазмы.
Уменьшению миотонических проявлений способствует прием хинина, прокаинамида, фенитоина в сочетании с ацетазоламидом. Показаны анаболические стероиды ( нандролона деканоат, метиландростендиол, метандростенол), небольшие дозы АТФ, витамины группы В.
Источник: https://www.KrasotaiMedicina.ru/diseases/zabolevanija_neurology/rossolimo-steinert-curschmann
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — это заболевание наследственного характера, прогрессирование которого происходит чрезвычайно медленными темпами. Данное нарушение наследуется только по аутосомно-доминантному типу, что происходит крайне редко — частота случаев составляет от 2 пациентов до 5 на 100 000 населения.
Содержание статьи:
Заболевание является мультисистемным и характеризуется вариабельной пенетрантностью патологического гена. Сопутствующими признаками являются аритмия, патологии эндокринной системы, ухудшение интеллекта. Заболевание классифицируется на два типа: врожденный и классический.
Причины развития миотонии Россолимо-Штейнерта-Куршмана
Миотония Россолимо-Штейнерта-Куршмана — это всегда генетически передающийся порок. Ключевой причиной его развития считается нарушение в гене DMPK, размещенном в девятнадцатой хромосоме.
Тяжесть течения заболевания прямо пропорциональна количеству тринуклеотидных CTG.
В норме этот показатель приблизительно равен 5-37, при 50-80 наблюдается мягкая форма заболевания, при 100-500 — поздняя тяжелая форма.
Под влиянием нарушения DMPK-гена в организме происходит изменение миотонинпротеинкиназы — белка, локализующегося в скелетной и мышечной ткани, в миокарде, ЦНС и др.
, что провоцирует появление миотонических спазмов, сочетающихся с атрофией мышц лица, шеи и конечностей в дистальных зонах. По сути, происходит гипертрофия одних волокон мышц с одновременной атрофией других.
В результате часть этих волокон замещается либо другими волокнами, либо жировыми или соединительными тканями.
Симптомы и признаки классической миотонии Россолимо-Штейнерта-Куршмана
Самые первые признаки миотонии могут проявляться уже в возрасте 6-7 лет, но чаще всего становятся хорошо заметны в подростковом и молодом возрасте от 10 до 20 лет. В перечне данных признаков присутствует миопатия, катаракта, поражения ЦНС и сердечно-сосудистой системы, эндокринные нарушения.
Основной симптом — наличие мышечных спазмов, при которых главным образом затрагивается жевательная лицевая мышца и мышца-сгибатель кисти. Также специалисты всегда отмечают атрофию различных мышц, включая дистальные отделы всех конечностей, височные, грудино-ключично-сосцевидные, мимические.
Наблюдается миопатический парез гортани с затруднением дыхания, изменениями голоса, ухудшением вентиляции легких, патологиями сна и аспирационной и застойной пневмонией.
Из-за изменений в мышечных тканях и преждевременного угасания рефлексов сухожилий у большинства пациентов происходит изменение походки.
Примерно в 50% случаев больные жалуются на такой симптом, как аритмия, наряду с которой могут наблюдаться блокада ножек пучка Гиса, а также гипертрофия левого сердечного желудочка.
Со стороны СНС заболевание вызывает гиперсомнию, снижение интеллектуальных способностей, нередки случаи, когда у больных отмечалась легкая форма дебильности.
Из-за патологий эндокринной системы, происходят нарушения половой сферы.
У молодых женщин изменения состоят в проявлении гирсутизма, сбоях менструального цикла и раннем климаксе, у молодых людей — в снижении или отсутствии влечения и импотенции, крипторхизме и гипогонадизме.
У представителей обоих полов имеется общий симптом миотонии Россолимо-Штейнерта-Куршмана — это изменения структуры волос с их последующей потерей. У мужчин облысение происходит в зоне висков и лба, у женщин проявляется местное очаговое или диффузное нарушение.
Симптомы миотонии Россолимо-Штейнерта-Куршмана врожденного типа
Врожденный тип миотонии может быть заметен специалистом еще в период пребывания в материнской утробе. Такой плод не проявляет повышенной двигательной активности. Поэтому если беременная женщина замечает подобное поведение малыша в животе, ей следует в обязательном порядке пройти УЗИ в последние три месяца перед родами.
Признаки миотонии присутствуют и у новорожденных. Это гипотония мимических, жевательных мышц, дистальных зон конечностей, глазных яблок. Кроме того, нередки и расстройства дыхания.
Немного позже проявляются такие симптомы, как олигофрения и задержка моторного развития. Прогрессирование этих патологий может происходить с разной скоростью.
При быстрых темпах их развития смерть пациента от заболевания может постичь его уже в самом раннем возрасте.
Диагностика миотонии Россолимо-Штейнерта-Куршмана
Подозрения специалиста на данное заболевание может вызвать сочетание любых миотонических проявлений и дистрофических изменений в тканях мышц, происходящих одновременно с отставанием в интеллектуальном развитии, нарушениями деятельности эндокринной и сердечно-сосудистой системы.
Наличие у больного всех этих признаков требует подтверждения диагноза в виде результатов проведения генеалогического анализа, результаты которого говорят об аутосомно-доминантном типе генетического наследования. Кроме них для точного диагностирования следует получить данные анализа ДНК.
Необходимы также дополнительные исследования: электромиография, электронейрография, электрокардиограмма, анализ гормонального фона.
Выяснение точного диагноза при подозрении на наличие миотонии Россолимо-Штейнерта-Куршмана требует всестороннего изучения состояния пациента и работы практически всех его органов. Поэтому к процессу диагностирования могут быть привлечены специалисты по генетике, кардиологии, гинекологии, андрологии и эндокринологи.
При диагностировании нужно обращать внимание не только на те изменения, которые проявляются внешне и являются характерными для миотонии Россолимо-Штейнерта-Куршмана, но и на данные по биопсии мышц и электромиографическому исследованию.
Во время проведения биохимического анализа изучается мышечный фермент и его уровень в тканях, который при данном заболевании всегда повышен. Обязательным условием постановки точного диагноза является проведение антенатальной диагностики миотонии методом амниоцентеза.
Дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршмана
Необходимо дифференцировать миотонию Россолимо-Штейнерта-Куршмана от прочих известных видов миотонии, которые в ряде ситуаций могут иметь схожие признаки и течение.
Атрофия мышечных тканей характерна исключительно для этого заболевания и позволяет быстро отличить его от миотонии Томсена, признаком которой является гипертрофия мышц. Раннее поражение лицевых мышц и доминантное наследование отличают его от миотонии Беккера.
Помимо указанных состояний, необходимо дифференцировать заболевание Россолимо-Штейнерта-Куршмана от БАС и амиотрофии Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
В современной медицинской практике в настоящее время нет терапии, предназначенной для полного излечения людей от миотонии Россолимо-Штейнерта-Куршмана.
Пациентам, у которых обнаружено это заболевание, назначают диету, основу которой составляют продукты с минимальным уровнем содержания калия или вовсе без него.
Людям, страдающим данной миотонией, не следует переохлаждаться, так как низкие температуры могут в любом момент спровоцировать спазмы.
Для уменьшения риска их внезапного появления и улучшения общего состояния больного, ему назначают следующие препараты: хинин, новокаинамид, дифенин одновременно с диакарбом.
К этим медикаментам специалисты рекомендуют добавить обязательный прием анаболических стероидов: неробола, ретаболила, метиландростендиола.
Необходимо также включить в список обязательных для приема веществ небольшие дозы АТФ и витамины группы В.
Указанные выше препараты оказывают хороший эффект как при классическом, так и при врожденном типе заболевания. Все перечисленные меры позволяют снизить негативные влияния заболевания на организм пациента и продлить срок его жизни, однако они никак не способны полностью избавить его от миотонии и снова сделать абсолютно здоровым человеком.
Источник: https://www.mosmedportal.ru/illness/distroficheskaya-miotoniya-rossolimo-shteynerta-kurshmana/
Дистрофическая миотония Россолимо-Штейнерта-Куршмана: причины заболевания, основные симптомы, лечение и профилактика
Медленно прогрессирующая генетическая болезнь, которая выражается дефектом миотонин-потеинкиназы. В результате развивается миотония и дистрофические изменения мышц.
Основными симптомами являются миотонические спазмы, атрофические изменения лицевых и шейных мышц, снижение интеллектуальной функции, аритмия, эндокринная патология, атрофия мускулов дистальных отделов рук и ног.
Заболевание диагностируется неврологом, но иногда пациента также консультируют генетик, кардиолог, эндокринолог, гинеколог и андролог. Врач анализирует симптомы, изучает историю болезни проводит неврологический осмотр и назначает дополнительные обследования.
Как правило, выполняют генеалогический анализ, исследование ДНК, электромиографию, электронейрографию, исследование гонадостероидов и электрокардиографию. Лечение направлено на купирование симптоматики. Больным назначат прием Фенитоина, Прокаинамида, Хинина, мочегонных препаратов, анаболических стероидов и аденозинтрифосфатной кислоты. Недуг имеет аутосомно-доминантный тип передачи.
Причины миотонии Россолимо-Штейнерта-Куршмана
Заболевание развивается на фоне дефекта гена DMPK, расположенного в 19-й хромосоме, который отвечает за синтез данного подкласса ферментов киназ.
Обычно, тринуклеотидные CTG-повторы значительно увеличиваются в основной части дефектного гена. Тип недуга и выраженность симптоматики напрямую связана с количеством повторов.
Если мутации гена передались ребенку от матери, могут возникать тяжелые формы болезни либо, она проявится сразу после рождения.
Симптомы миотонии Россолимо-Штейнерта-Куршмана
Первые симптомы начинают проявляться в дошкольном возрасте или могут дебютировать до 35 лет. Чаще всего признаки недуга выявляются между десятым и двадцатым годом жизни. У пациентов наблюдаются длительные тонические спазмы мышц, слабость в определённых группах мышц, поражения сердечно-сосудистой и центральной нервной систем, эндокринные нарушения и катаракта.
Миотонические спазмы интенсивнее всего поражают жевательные мышцы и мышцы-сгибатели кистей. Когда невролог проверяет рефлексы неврологическим молотком, отмечается механическая миотоническая реакция. Также заболевание проявляется атрофическими изменениями, поражающими разные группы мышц.
По мере развития недуга мышечная дистрофия прогрессирует, а миотонические спазмы угасают.
Наиболее часто спазмы отмечаются в мышцах дистальных отделов рук и ног, мимической мускулатуре, грудино-ключично-сосцевидных и височных мышцах. При поражении лицевых мышц у пациента может образоваться маскообразное печальное выражение лица.
На фоне атрофии мышц гортани или глотки развиваются миопатический парез гортани, затруднения акта глотания, а также нарушения голоса.
В некоторых случаях атрофируются или спазмируются дыхательные мышцы, что может выражаться ухудшением вентиляции легких, приступами отсутствия дыхания во сне, развитием таких заболеваний, как застойная или аспирационная пневмония.
Около 50% пациентов страдают от патологий сердечно-сосудистой системы: аритмиями, блокадой ножек пуска Гиса, гипертрофией левого желудочка. При нарушениях центральной нервной системы интеллектуальные функции снижаются, а также наблюдается чрезмерная дневная сонливость.
Кроме того, у некоторых больных отмечаются расстройства эндокринной системы: снижение либидо, крипторхизм, импотенция, гипогонадизм, гирсутизм, нарушения менструации, ранний климакс, алопеция.
При врожденном типе недуга его признаки можно обнаружить с помощью акушерского ультразвукового исследования после 28 недели беременности.
Диагностика миотонии Россолимо-Штейнерта-Куршмана
Заболевание диагностируется неврологом, но иногда пациента также консультируют генетик, кардиолог, эндокринолог, гинеколог и андролог.
Врач анализирует симптомы, изучает историю болезни проводит неврологический осмотр и назначает дополнительные обследования.
Как правило, выполняют генеалогический анализ, исследование ДНК, электромиографию, электронейрографию, исследование гонадостероидов и электрокардиографию.
Лечение миотонии Россолимо-Штейнерта-Куршмана
Специалисты проводят симптоматическую терапию. Больным регулируют рацион питания, из которого исключают продукты с повышенным содержанием калия. Чтобы уменьшить миотонические проявления назначают: Хинин, Прокаинамид, Фенитоин, Ацетазоламид, Метиландростендиол, Метандростенол, Аденозинтрифосфатную кислоту, и витамин В.
Профилактика миотонии Россолимо-Штейнерта-Куршмана
Специфические методы профилактики не разработаны. Парам, планирующим беременность, рекомендовано посетить консультацию генетика.
Источник: https://www.obozrevatel.com/health/bolezni/distroficheskaya-miotoniya-rossolimo-shtejnerta-kurshmana.htm






